Atomic And Molecular Clusters Latest Preprints | 2019-04-09
Atomic And Molecular Clusters
Few-body quantum method in a 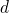 -dimensional space (1901.02667v2)
-dimensional space (1901.02667v2)
E. Garrido, A. S. Jensen, R. Álvarez-Rodríguez
2019-01-09
In this work we investigate the continuous confinement of quantum systems from three to two dimensions. Two different methods will be used and related. In the first one the confinement is achieved by putting the system under the effect of an external field. This method is conceptually simple, although, due to the presence of the external field, its numerical implementation can become rather cumbersome, especially when the system is highly confined. In the second method the external field is not used, and it simply considers the spatial dimension
A tight-binding model for the excitonic band structure of a one-dimensional molecular chain: UV-Vis spectra, Zak phase and topological properties (1904.01881v1)
Wei Wu, Jin Zhang
2019-04-03
Recently organic optics becomes a hot topic due to the rapid development of organic light-emitting diodes, organic solar cells, and organic photon detectors. The optical spectra of the molecular semiconductors are difficult to solve an model from first-principles because (i) the very large number of atoms in a unit cell and (ii) the accurate theoretical excited state is still under development. Here we present a tight-binding model of an exciton band structure in a molecular chain. We take into account the intra-molecule and charge-transfer excitation within a molecular dimer in a unit cell, then we apply the tight-binding model by including the coupling between two types of excitations. We not only found that our calculations can explain a body of UV-Vis optical spectra of transition-metal phthalocyanines, but also a one-dimensional excitonic topological band structure if we fine-tune the couplings in a dimerized molecular chain. We have found a large space to obtain the topological Zak phase in the parameter space, in which there is a simple linear relationship between the hopping integrals between cells and within cell.
Stretched or noded orbital densities and self-interaction correction in density functional theory (1903.00611v2)
Chandra Shahi, Puskar Bhattarai, Kamal Wagle, Biswajit Santra, Sebastian Schwalbe, Torsten Hahn, Jens Kortus, Koblar A. Jackson, Juan E. Peralta, Kai Trepte, Susi Lehtola, Niraj K. Nepal, Hemanadhan Myneni, Bimal Neupane, Santosh Adhikari, Adrienn Ruzsinszky, Yoh Yamamoto, Tunna Baruah, Rajendra R. Zope, John P. Perdew
2019-03-02
Semi-local approximations to the density functional for the exchange-correlation energy of a many-electron system necessarily fail for lobed one-electron densities, including not only the familiar stretched densities but also the less familiar but closely-related noded ones. The Perdew-Zunger (PZ) self-interaction correction (SIC) to a semi-local approximation makes that approximation exact for all one-electron ground- or excited-state densities and accurate for stretched bonds. When the minimization of the PZ total energy is made over real localized orbitals, the orbital densities can be noded, leading to energy errors in many-electron systems. Minimization over complex localized orbitals yields nodeless orbital densities, which reduce but typically do not eliminate the SIC errors of atomization energies. Other errors of PZ SIC remain, attributable to the loss of the exact constraints and appropriate norms that the semi-local approximations satisfy, and suggesting the need for a generalized SIC. These conclusions are supported by calculations for one-electron densities, and for many-electron molecules. While PZ SIC raises and improves the energy barriers of standard generalized gradient approximations (GGA's) and meta-GGA's, it reduces and often worsens the atomization energies of molecules. Thus PZ SIC raises the energy more as the nodality of the valence localized orbitals increases from atoms to molecules to transition states. PZ SIC is applied here in particular to the SCAN meta-GGA, for which the correlation part is already self-interaction-free. That property makes SCAN a natural first candidate for a generalized SIC.
Universal Three-Body Parameter of Heavy-Heavy-Light systems with negative intraspecies scattering length (1903.09565v1)
Caiyun Zhao, Huili Han, Mengshan Wu, Tingyun Shi
2019-03-22
The Three-Body Parameter(3BP)
is crucial to understanding Efimov physics, and a universal 3BP has been shown in experiments and theory in ultracold homonuclear gases. The 3BP of heteronuclear systems was predicted to possess much richer properties than the homonuclear counterparts for the large parameter space. In this work, we investigate the universal properties of
. We find that
. In a special case, when the two heavy atoms are in resonance,
.
The CO2-broadened H2O continuum in the 100-1500 cm-1 region. Measurements, predictions and empirical model (1903.08972v1)
Ha Tran, Martin Turbet, Simon Hanoufa, Xavier Landsheere, Pascale Chelin, Qiancheng Ma, Jean-Michel Hartmann
2019-03-21
Transmission spectra of H
O+CO
. The results are in good agreement with those, around 1300 cm
calculated, almost thirty years ago, using a quasistatic approach and an input H
Embedding nuclear physics inside the unitary window (1903.08900v1)
Mario Gattobigio, Alejandro Kievsky, Michele Viviani
2019-03-21
The large values of the singlet and triplet scattering lengths locate the two-nucleon system close to the unitary limit, the limit in which these two values diverge. As a consequence, the system shows a continuous scale invariance which strongly constrains the values of the observables, a well-known fact already noticed a long time ago. The three-nucleon system shows a discrete scale invariance that can be observed by correlations of the triton binding energy with other observables as the doublet nucleon-deuteron scattering length or the alpha-particle binding energy. The low-energy dynamics of these systems is universal; it does not depend on the details of the particular way in which the nucleons interact. Instead, it depends on a few control parameters, the large values of the scattering lengths and the triton binding energy. Using a potential model with variable strength set to give values to the control parameters, we study the spectrum of
nuclei in the region between the unitary limit and their physical values. In particular, we analyze how the binding energies emerge from the unitary limit forming the observed levels.
Single-atom control of the optoelectronic response in sub-nanometric cavities (1903.08443v1)
Pablo Garcia-Gonzalez, Alejandro Varas, F. J. Garcia-Vidal, Angel Rubio
2019-03-20
By means of ab-initio time dependent density functional theory calculations carried out on an prototypical hybrid plasmonic device (two metallic nanoparticles bridged by a one-atom junction), we demonstrate the strong interplay between photoinduced excitation of localized surface plasmons and electron transport through the single atom. Such an interplay is remarkably sensitive to the atomic orbitals of the junction. Therefore, we show the possibility of a twofold tuning (plasmonic response and photoinduced current across the juntion) just by changing a single atom in the device.
Accurate Electron Affinities and Orbital Energies of Anions from a Non-Empirically Tuned Range-Separated Density Functional Theory Approach (1803.01218v2)
Lindsey N. Anderson, M. Belén Oviedo, Bryan M. Wong
2018-03-03
The treatment of atomic anions with Kohn-Sham density functional theory (DFT) has long been controversial since the highest occupied molecular orbital (HOMO) energy,
, is often calculated to be positive with most approximate density functionals. We assess the accuracy of orbital energies and electron affinities for all three rows of elements in the periodic table (H-Ar) using a variety of theoretical approaches and customized basis sets. Among all of the theoretical methods studied here, we find that a non-empirically tuned range-separated approach (constructed to satisfy DFT-Koopmans' theorem for the anionic electron system) provides the best accuracy for a variety of basis sets - even for small basis sets where most functionals typically fail. Previous approaches to solve this conundrum of positive
Simulating the structural diversity of carbon clusters across the planar to fullerene transition (1902.09593v2)
Maëlle A. Bonnin, Cyril Falvo, Florent Calvo, Thomas Pino, Pascal Parneix
2019-02-25
Together with the second generation REBO reactive potential, replica-exchange molecular dynamics simulations coupled with systematic quenching were used to generate a broad set of isomers for neutral C
clusters with
, 42, and 60. All the minima were sorted in energy and analyzed using order parameters to monitor the evolution of their structural and chemical properties. The structural diversity measured by the fluctuations in these various indicators is found to increase significantly with energy, the number of carbon rings, especially 6-membered, exhibiting a monotonic decrease in favor of low-coordinated chains and branched structures. A systematic statistical analysis between the various parameters indicates that energetic stability is mainly driven by the amount of sp
hybridization, more than any geometrical parameter. The astrophysical relevance of these results is discussed in the light of the recent detection of C
and C
fullerenes in the interstellar medium.
User Manual for MOLSCAT, BOUND and FIELD: programs for quantum scattering properties and bound states of interacting pairs of atoms and molecules (1903.06755v1)
Jeremy M. Hutson, C. Ruth Le Sueur
2019-03-15
MOLSCAT is a general-purpose package for performing non-reactive quantum scattering calculations for atomic and molecular collisions using coupled-channel methods. Simple atom-molecule and molecule-molecule collision types are coded internally and additional ones may be handled with plug-in routines. Plug-in routines may include external magnetic, electric or photon fields (and combinations of them). Simple interaction potentials are coded internally and more complicated ones may be handled with plug-in routines. BOUND is a general-purpose package for performing calculations of bound-state energies in weakly bound atomic and molecular systems using coupled-channel methods. It solves the same sets of coupled equations as \MOLSCAT, and can use the same plug-in routines if desired, but with different boundary conditions. FIELD is a development of BOUND that locates external fields at which a bound state exists with a specified energy. One important use is to locate the positions of magnetically tunable Feshbach resonance positions in ultracold collisions. Previous versions of these programs have been released separately. However, there is a significant degree of overlap between their internal structures and usage specifications. This manual therefore describes all three, with careful identification of parts that are specific to one or two of the programs.