Atomic And Molecular Clusters Latest Preprints | 2019-03-12
Atomic And Molecular Clusters
ZMP-SAPT: DFT-SAPT using ab initio Densities (1903.03350v1)
A. Daniel Boese, Georg Jansen
2019-03-08
Symmetry Adapted Perturbation Theory (SAPT) has become an important tool when predicting and analyzing intermolecular interactions. Unfortunately, DFT-SAPT, which uses Density Functional Theory (DFT) for the underlying monomers, has some arbitrariness concerning the exchange-correlation potential and the exchange-correlation kernel involved. By using ab initio Brueckner Doubles densities and constructing Kohn-Sham orbitals via the Zhao-Morrison-Parr (ZMP) method, we are able to lift the dependence of DFT-SAPT on DFT exchange-correlation potential models in first order. This way, we can compute the monomers at the Coupled-Cluster level of theory and utilize SAPT for the intermolecular interaction energy. The resulting ZMP-SAPT approach is tested for small dimer systems involving rare gas atoms, cations, and anions and shown to compare well with the Tang-Toennies model and coupled cluster results.
Deep neural networks for classifying complex features in diffraction images (1903.02779v1)
Julian Zimmermann, Bruno Langbehn, Riccardo Cucini, Michele Di Fraia, Paola Finetti, Aaron C. LaForge, Toshiyuki Nishiyama, Yevheniy Ovcharenko, Paolo Piseri, Oksana Plekan, Kevin C. Prince, Frank Stienkemeier, Kiyoshi Ueda, Carlo Callegari, Thomas Möller, Daniela Rupp
2019-03-07
Intense short-wavelength pulses from free-electron lasers and high-harmonic-generation sources enable diffractive imaging of individual nano-sized objects with a single x-ray laser shot. The enormous data sets with up to several million diffraction patterns represent a severe problem for data analysis, due to the high dimensionality of imaging data. Feature recognition and selection is a crucial step to reduce the dimensionality. Usually, custom-made algorithms are developed at a considerable effort to approximate the particular features connected to an individual specimen, but facing different experimental conditions, these approaches do not generalize well. On the other hand, deep neural networks are the principal instrument for today's revolution in automated image recognition, a development that has not been adapted to its full potential for data analysis in science. We recently published in Langbehn et al. (Phys. Rev. Lett. 121, 255301 (2018)) the first application of a deep neural network as a feature extractor for wide-angle diffraction images of helium nanodroplets. Here we present the setup, our modifications and the training process of the deep neural network for diffraction image classification and its systematic benchmarking. We find that deep neural networks significantly outperform previous attempts for sorting and classifying complex diffraction patterns and are a significant improvement for the much-needed assistance during post-processing of large amounts of experimental coherent diffraction imaging data.
Interactions of benzene, naphthalene, and azulene with alkali-metal and alkaline-earth-metal atoms for ultracold studies (1903.01378v1)
Paweł Wójcik, Tatiana Korona, Michał Tomza
2019-03-04
We consider collisional studies of polyatomic aromatic hydrocarbon molecules immersed into ultracold atomic gases and investigate intermolecular interactions of exemplary benzene, naphthalene, and azulene with alkali-metal (Li, Na, K, Rb, Cs) and alkaline-earth-metal (Mg, Ca, Sr, Ba) atoms. We apply the state-of-the-art ab initio techniques to compute the potential energy surfaces (PESs). We use the coupled cluster method restricted to single, double, and noniterative triple excitations to reproduce the correlation energy and the small-core energy-consistent pseudopotentials to model the scalar relativistic effects in heavier metal atoms. We also report the leading long-range isotropic and anisotropic dispersion and induction interaction coefficients. The PESs are characterized in detail and the nature of intermolecular interactions is analyzed and benchmarked using symmetry-adapted perturbation theory. The full three-dimensional PESs are provided for selected systems within the atom-bond pairwise additive representation and can be employed in scattering calculations. Presented study of the electronic structure is the first step towards the evaluation of prospects for sympathetic cooling of polyatomic aromatic molecules with ultracold atoms. We suggest azulene, an isomer of naphthalene which possesses a significant permanent electric dipole moment and optical transitions in the visible range, as a promising candidate for electric field manipulation and buffer-gas or sympathetic cooling.
Stretched or noded orbital densities and self-interaction correction in density functional theory (1903.00611v1)
Chandra Shahi, Puskar Bhattarai, Kamal Wagle, Biswajit Santra, Sebastian Schwalbe, Torsten Hahn, Jens Kortus, Koblar A. Jackson, Juan E. Peralta, Kai Trepte, Susi Lehtola, Niraj K. Nepal, Hemanadhan Myneni, Bimal Neupane, Santosh Adhikari, Adrienn Ruzsinszky, Yoh Yamamoto, Tunna Baruah, Rajendra R. Zope, John P. Perdew
2019-03-02
Semi-local approximations to the density functional for the exchange-correlation energy of a many-electron system necessarily fail for lobed one-electron densities, including not only the familiar stretched densities but also the less familiar but closely-related noded ones. The Perdew-Zunger (PZ) self-interaction correction (SIC) to a semi-local approximation makes that approximation exact for all one-electron ground- or excited-state densities and accurate for stretched bonds. When the minimization of the PZ total energy is made over real localized orbitals, the orbital densities can be noded, leading to energy errors in many-electron systems. Minimization over complex localized orbitals yields nodeless orbital densities, which reduce but typically do not eliminate the SIC errors of atomization energies. Other errors of PZ SIC remain, attributable to the loss of the exact constraints and appropriate norms that the semi-local approximations satisfy, and suggesting the need for a generalized SIC. These conclusions are supported by calculations for one-electron densities, and for many-electron molecules. While PZ SIC raises and improves the energy barriers of standard generalized gradient approximations (GGA's) and meta-GGA's, it reduces and often worsens the atomization energies of molecules. Thus PZ SIC raises the energy more as the nodality of the valence localized orbitals increases from atoms to molecules to transition states. PZ SIC is applied here in particular to the SCAN meta-GGA, for which the correlation part is already self-interaction-free. That property makes SCAN a natural first candidate for a generalized SIC.
Observation of highly dispersive bands in pure thin film C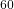 (1902.10827v1)
(1902.10827v1)
Drew W. Latzke, Claudia Ojeda-Aristizabal, Sinéad M. Griffin, Jonathan D. Denlinger, Jeffrey B. Neaton, Alex Zettl, Alessandra Lanzara
2019-02-27
While long-theorized, the direct observation of multiple highly dispersive C
Se
--through the use of angle-resolved photoemission spectroscopy (ARPES) and first-principles calculations. The effects of this novel substrate reducing C
Time-resolved observation of interatomic Coulombic decay induced by two-photon double excitation of Ne (1902.09882v1)
T. Takanashi, N. V. Golubev, C. Callegari, H. Fukuzawa, K. Motomura, D. Iablonskyi, Y. Kumagai, S. Mondal, T. Tachibana, K. Nagaya, T. Nishiyama, K. Matsunami, P. Johnsson, P. Piseri, G. Sansone, A. Dubrouil, M. Reduzzi, P. Carpeggiani, C. Vozzi, M. Devetta, M. Negro, D. Faccialà, F. Calegari, A. Trabattoni, M. C. Castrovilli, Y. Ovcharenko, M. Mudrich, F. Stienkemeier, M. Coreno, M. Alagia, B. Schütte, N. Berrah, O. Plekan, P. Finetti, C. Spezzani, E. Ferrari, E. Allaria, G. Penco, C. Serpico, G. De Ninno, B. Diviacco, S. Di Mitri, L. Giannessi, G. Jabbari, K. C. Prince, L. S. Cederbaum, Ph. V. Demekhin, A. I. Kuleff, K. Ueda
2019-02-26
The hitherto unexplored two-photon doubly-excited states [Ne
(
)]
ions was recorded as a function of delay between the XUV pump and UV probe laser pulses. The extracted lifetimes of the long-lived doubly-excited states, 390 (-130 / +450} fs, and of the short-lived ones, less than 150~fs, are in good agreement with \emph{ab initio} quantum mechanical calculations.
Simulating the structural diversity of carbon clusters across the planar to fullerene transition (1902.09593v1)
Maëlle A. Bonnin, Cyril Falvo, Florent Calvo, Thomas Pino, Pascal Parneix
2019-02-25
Together with the second generation REBO reactive potential, replica-exchange molecular dynamics simulations coupled with systematic quenching were used to generate a broad set of isomers for neutral C
clusters with
, 42, and 60. All the minima were sorted in energy and analyzed using order parameters to monitor the evolution of their structural and chemical properties. The structural diversity measured by the fluctuations in these various indicators is found to increase significantly with energy, the number of carbon rings, especially 6-membered, exhibiting a monotonic decrease in favor of low-coordinated chains and branched structures. A systematic statistical analysis between the various parameters indicates that energetic stability is mainly driven by the amount of sp
hybridization, more than any geometrical parameter. The astrophysical relevance of these results is discussed in the light of the recent detection of C
fullerenes in the interstellar medium.
Magic Numbers for the Photoelectron Anisotropy in Li-Doped Dimethyl Ether Clusters (1902.09575v1)
Jonathan V. Barnes, Bruce L. Yoder, Ruth Signorell
2019-02-25
Photoelectron velocity map imaging of Li(CH
OCH
n
Propensity rules in photoelectron circular dichroism in chiral molecules I: Chiral hydrogen (1806.09049v2)
Andres F. Ordonez, Olga Smirnova
2018-06-23
Photoelectron circular dichroism results from one-photon ionization of chiral molecules by circularly polarized light and manifests itself in forward-backward asymmetry of electron emission in the direction orthogonal to the light polarization plane. What is the physical mechanism underlying asymmetric electron ejection? How "which way" information builds up in a chiral molecule and maps into forward-backward asymmetry? We introduce instances of bound chiral wave functions resulting from stationary superpositions of states in a hydrogen atom and use them to show that the chiral response in one-photon ionization of aligned molecular ensembles originates from two propensity rules: (i) Sensitivity of ionization to the sense of electron rotation in the polarization plane. (ii) Sensitivity of ionization to the direction of charge displacement or stationary current orthogonal to the polarization plane. In the companion paper we show how the ideas presented here are part of a broader picture valid for all chiral molecules and arbitrary degrees of molecular alignment.
Prediction of activation energy barrier of island diffusion processes using data-driven approaches (1902.10282v1)
Shree Ram Acharya, Talat S. Rahman
2019-02-22
We present models for prediction of activation energy barrier of diffusion process of adatom (1-4) islands obtained by using data-driven techniques. A set of easily accessible features, geometric and energetic, that are extracted by analyzing the variation of the energy barriers of a large number of processes on homo-epitaxial metallic systems of Cu, Ni, Pd, and Ag are used along with the activation energy barriers to train and test linear and non-linear statistical models. A multivariate linear regression model trained with energy barriers for Cu, Pd, and Ag systems explains 92% of the variation of energy barriers of the Ni system, whereas the non-linear model using artificial neural network slightly enhances the success to 93%. Next mode of calculation that uses barriers of all four systems in training, predicts barriers of randomly picked processes of those systems with significantly high correlation coefficient: 94.4% in linear regression model and 97.7% in artificial neural network model. Calculated kinetics parameters such as the type of frequently executed processes and effective energy barrier for Ni dimer and trimer diffusion on the Ni(111) surface obtained from KMC simulation using the predicted (data-enabled) energy barriers are in close agreement with those obtained by using energy barriers calculated from interatomic interaction potential.