Iron Overload In Beta-Thalassemia Patient
Today, when I was on a clinic duty, one patient came to me for a routine follow up. He has been attending so many appointments with the paediatric clinic since he was diagnosed with thalassemia major (Beta thalassemia) since he was a child. Now, he is 26 years old, and believe me, when I said, he doesn't look like one. When he walked towards me, I thought he was somewhere around 15-18 years old with his short stature, darkly pigmented skin and slightly overweight, but it turns out, he is of the same age as me. His physical appearances can be attributed to the complications of having a prolonged blood transfusion for his condition.
Thalassemia can be inherited from affected parents to their child which would usually be presented with lethargy, anaemia, jaundice and even stunted growth. The affected child would inherit a condition which would render the haemoglobin production defective as a result of genetic mutation. Alteration of a set of genes which controls the globin chain production would cause the unaffected globin chain to accumulate within the cell membrane of the red blood cells making it ineffective in carrying out its primary function. They would either:
- Die inside the bone marrow (ineffective erythropoiesis)
- Being removed from the circulation prematurely ( short lifespan of red blood cells)
Either way, people with thalassemia would have a hypochromic microcytic appearance of the red blood cells (it's small and dilute compared to the other healthy red blood cells). There are a few types of haemoglobin which can be considered healthy if it is present at an appropriate level in the circulation:
- Hb A: This type of haemoglobin makes up for 97% of the total adult haemoglobins. It comprises the combination of two alpha protein chains and two beta protein chains.
- Hb A1c: This is a glycosylated haemoglobin. In a normal individual, HbA1c stays below 5% of the total haemoglobins. An increased level of HbA1c is associated with poor healing and multiple complications which can usually be observed in a diabetic patient. It is formed when a haemoglobin (Hb A) bond with the circulating glucose in a harmful reaction called glycation.

- Hb A2: This particular haemoglobin type makes up for 2% of the total adult haemoglobin. Due to the disease process in Beta-thalassemia, Hb A2 might increase beyond its expected level to compensate for the reduced production of viable Hb A. It comprises the combination of two alpha protein chains and two delta protein chains.
- Hb F: This is the primary haemoglobin type in a growing foetus which has a high affinity towards oxygen. It increased in a patient with Thalassemia to compensate for the reduced production of viable Hb A.
In the course of humans development, the first kind of haemoglobin which supplies for major organ of a foetus during the first trimester were Hb Gower, Hb Portland and Hb F. They are the products of a different combination of globin chains such as Epsilon, Alpha, Gamma and Zeta. Beta chains were produced since the early development of a foetus in the first trimester but their production was kept at a low level. When the foetus reaches the third trimester, only then, the production of beta chain increased significantly, preparing the foetus for the life outside of its mother's womb.
The predominant type of haemoglobin during the intrauterine life is the Hb F. They provide oxygen to the foetus and will continue to do so until it becomes scarce, 6 months after delivery. At that point of time, the role of Hb F would be taken primarily by Hb A and Hb A2. The sequence is quite simple:
- Hb Gower, Hb Portland (First trimester)
- Hb F (From the first trimester until birth)
- Hb A and Hb A2 (After 6 months postpartum)

A patient who has been diagnosed with thalassemia will either produce less beta chain or less alpha chain. As we can see, we need alpha chain since the beginning of the first trimester, so logically, if a patient presented with alpha thalassemia, they will show signs and symptoms of anaemia early. People with beta-thalassemia were usually diagnosed approximately after 3 months of life. The onset can be later than that due to the fact that, the role of carrying oxygen can still be carried out by Hb F and Hb A2 during the first 6 months of life making them symptom-free. It will, however, depends on the type of beta thalassemia they were having.
| Beta Thalassemia Minor | They can be asymptomatic for life. Blood investigations can reveal a mild form of anaemia with a slight increase of Hb A2 and Hb F. They do not need a blood transfusion |
| Beta Thalassemia intermedia | Patient with this form of thalassemia would present with signs and symptoms indicating a moderate form of anaemia. They were usually diagnosed at the age of 2 years and don't necessarily need a blood transfusion. Some will need a blood transfusion during pregnancy or infection but if a patient keen for a blood transfusion due to poor growth rate or anything, the interval would vary from 4 to 12 weekly interval. |
| Beta Thalassemia major | Patients who are diagnosed with this type of thalassemia would have a homozygous pattern of inheritance. They will show some signs and symptoms related to anaemia as early as 3 months old making it difficult for them to achieve a necessary requirement for a healthy growth. Blood transfusions were necessary and at some point, they would have to be started on iron chelating agents as well. |
Blood Transfusion and Iron Overload
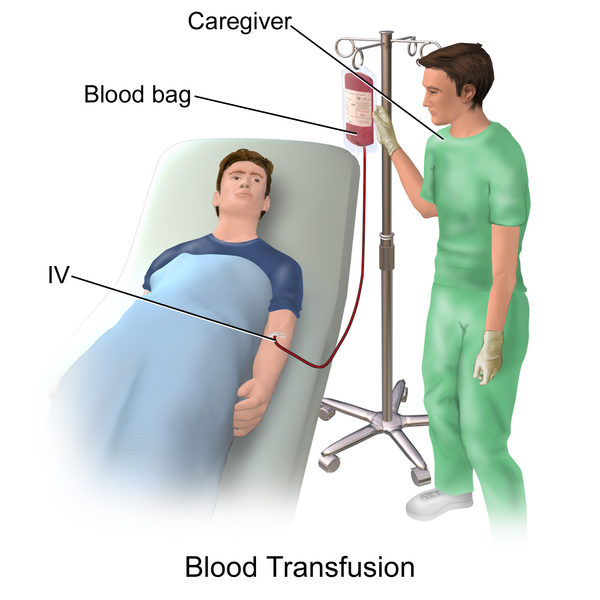
Blood transfusion has been the mainstay of treatment for patients who have been diagnosed with thalassemia. It is important to start transfusing blood as soon as:
- The diagnosis of major beta-thalassemia has been established by appropriate investigations
- The level of haemoglobin is less than 7 g/dl for more than a week in the absence of other relevant factors such as infection or pregnancy.
- A normal level of haemoglobin but there are other signs which indicate poor growth rate and extramedullary erythropoiesis such as maxillary bone overgrowth, hepatomegaly, splenomegaly and stunted growth.
An early blood transfusion will reduce the mortality rate among patient with beta-thalassemia but they are prone to one of the complications of blood transfusion which can affect the integrity of various organs in the body; iron overload. Iron overload can cause various complications depending on the site of iron deposition:
- If it affects the liver, patients will be presented with liver cirrhosis
- If it affects the pancreas, patients will be presented with diabetes mellitus
- If it affects the thyroid, patients will be presented with hypothyroidism and stunted growth
- If it affects the skin, patients will be presented with hyperpigmented skin

The level of haemoglobin decline is expected at approximately 1g/dl per week which would make it reasonable for a blood transfusion dependent thalassemia to come for blood transfusion once in every four weeks. In a normal human adult, we retain as much as 4g of iron in our body (3g is being kept in the blood while the rest is being stored in the liver). Every single day, we lose iron in a small amount via skin and gut but most of it will be replaced by our diet (10% of iron content in the diet will be absorbed by the gut). It is important to replace loss iron to prevent complications which can arise from iron deficiency and it is important to lose some iron to avoid iron overload.
Even though blood transfusion is the mainstay mode of treatment for patients with beta-thalassemia, they need to deal with complications of prolonged blood transfusion which is iron overload. Patients with the age of 3 years and above would have a serum ferritin approximately 1000 ng/ml (normal level is between 100-400 ng/ml) which can be considered a toxic level after 15-20 bouts of blood transfusion. Normally, this excess iron would be stored in the liver which explains why liver would be the first organ affected by iron overload. If the liver can't keep the excess iron, it will accumulate in various organs, damaging them.
.jpg){kind=link}
{kind=link}
{kind=link}
There are a few types of iron chelating agents which are commonly used to treat iron overload:
- Subcutaneous Desferrioxamine (Desferal)
- Oral Deferiprone (L1)
Desferal is delivered through a subcutaneous injection which can be painful for some patients. The pain factor is crucial as it can affect patient's adherence towards medication. It is important for a patient with thalassemia to become adherence in order to control the level of iron in the body. Desferral also can cause some local complications such as tender injection site, swelling, severe allergic reaction and Yersinia infection.
L1, on the other hand, is an oral medication which is much easier to take. It is usually prescribed if the effect provided by Desferal is sub-optimal or the patient is contraindicated from taking Desferal. It is much expensive and in a few cases, can cause a side effect called agranulocytosis. It can predispose the patient to a variety of infection as a consequence of leukopenia. Despite the side effect, people who are taking L1 were much more compliance compared to people who are taking Deferral.
In a peer review journal published in 2007 by Delea TE et al. has shown that the compliance rate of people who are taking deferral is much lower (between 58-78%) compared to people who are taking L1 (79-98%). Most of them have some issue with the pain inflicted by subcutaneous delivery of Deferral. It is important for a clinician to know which treatment is the best for the patient while taking into account their socioeconomic status and compliance rate. With a regular blood transfusion and iron chelating treatment, patients with beta-thalassemia major can live pretty close to the definition of a normal life.
Source
- The thalassemias and related disorders
- Iron Overload in Beta Thalassaemia Major and Intermedia Patients
- Blood transfusion among thalassemia patients: A single Egyptian center experience


SteemSTEM is a community project with the goal to promote and support Science, Technology, Engineering and Mathematics on the Steem blockchain. If you wish to support the steemSTEM project you can:
Contribute STEM content using the #steemstem tag | Support steemstem authors | Join our curation trail | Visit our Discord community | Delegate SP to steemstem
I really like your articles, and the way you bind them with actual cases you encounter is welcome. Sometimes I get overwhelmed by the info, I like to read though :P
You are doing great, and I see that SteemSTEM was even faster than me in reading and approving your article today :P
Keep it up and keep connecting here, your subjects are good and the writing is well structured.
Thanks, @alexdory. I appreciate your help. :)
Hi @conficker! Really sad to know about the patient's complications of having a prolonged blood transfusion for his conditions.
The article is quite interesting from which I came to know about many new facts. Thanks for bringing such a nice post! I loved the way in which the informations provided by you!!
Cheers!! 😇
No problem man. Glad you like it.
This is an excellent explanation of the challenges facing patients with Beta Thalassemia. Of course, those who merely carry the trait should have genetic counseling before having children, to make certain their partner does not also carry the trait. I understand there is a chance for those who carry the trait to have iron overload if they take iron supplements. Is this true?
The one thing that struck me in your article is that you mention a healthcare professional should consider socioeconomic status in recommending treatment options. This is a sad commentary on healthcare delivery (in the US, anyway). Optimal medical care should be based on optimal outcome, not on a patient's ability to pay.
Universal healthcare. In my view, the only moral choice, I believe.
Well, people who were diagnosed with Thalassemia are prone to get iron overload even without consuming iron supplement or blood transfusion. Patients were having ineffective red blood cells production which can lead to overabsorption of iron from the intestine. This is caused by some kind of misinterpretation by the body thinking that the anaemia was caused by iron depletion when in fact the iron level was normal.
Well, I agree but you should know that there are many factors which can contribute to the outcome of a treatment and one of them would be financial instability. Let say the price of the drugs were 500 USD per month and the patient salary is 1000 USD per month. He/she (the patient) would have other things to pay for (school fee, bills, rent) which if we add up with the cost of the medication can total up to more than 1000 USD per month. What do you think the patient would do? He might want to skip buying the medication until he has enough money to buy for another month supply.
This is an example of how financial instability affects compliance. Can that action be considered as an optimal health care? Nope. That's why it is important to consider the socioeconomic background of a patient before deciding on treatment modalities. In medicine, we are not only treating the disease but the patient as a whole (holistic approach). It will be unrealistic to treat patients with the most expensive kind of drug while leaving them with thousands of dollar of debt.
It's tough but as long as there is an alternative, it should be fine. The field of medicine is advancing day by day.
I agree with you. Now let me tell you something regarding the universal healthcare. In my country there are patients that receive state-funded medication that is over 1000-3000$ per month. Some of them (because are very poor) are selling the medications instead of taking them and choose to live with the pain, discomfort and with their life in danger so they could benefit from the resale of the medication, usually 10 or 20 times lower than the market price. State loses, people lose, black market sharks win. It's not that widespread but it's happening.
I am still 100% for Universal Healthcare. Just exposing the other side of the problem.
It is wrong, but I think I can understand why they were doing that. Sometimes it is difficult to fathom why certain things happen until we are all in the same boat as them.