Active and Inactive Protein Kinases: Structural Basis for Regulation
Introduction
Protein kinases and phosphatases play pivotal roles in regulating and coordinating aspects of metabolism, gene expression, cell growth, cell motility, cell differentiation, and cell division. As a result, if cellular life is to function in an orderly manner, the switching on and off of protein kinases and phosphatases is as crucial for their function as their catalytic activity.
The total number of distinct kinase domain sequences available is now approaching 400. Multiple sequence alignments have indicated that all protein kinases should have similar structures, and this has been confirmed by recent crystal structure determinations. Conserved features have been identified in 12 subdomain regions of all protein kinases, and residues from these subdomains have been implicated in essential roles in enzyme structure and function. Protein kinases exhibit variability in other parts of the kinase domain, and different kinases may contain additional domains, additional subunits, or both. These features allow several different mechanisms for control.
Control mechanisms that have been recognized to date include the following: control by additional subunits or domains that may function in response to second messengers (e.g., cyclic AMP binding to the regulatory subunit of cyclic AMP–dependent protein kinase [cAPK], Ca2+/calmodulin binding to calmodulin-dependent protein kinases, and Ca2+ and diacyl glyerol binding to N-terminal domains of protein kinase C); control by additional subunits whose level of expression varies depending on the functional state of the cell (e.g., cyclin regulation of the cyclin-dependent protein kinases [CDKs]); control by additional domains that target the kinase to different molecules or subcellular localizations (e.g., the SH2 and SH3 domains of the Src kinases); control by additional domains that inhibit the kinase activity by an autoregulatory process (e.g., myosin light chain kinase [MLCK]); and control by phosphorylation and dephosphorylation by kinases and phosphatases. Phosphorylation of specific threonine, serine, or tyrosine residues may occur at a number of sites. Some of these are located in the N-terminal or C-terminal portions of the polypeptide chain, which lie outside the kinase domain (e.g., in Src kinase and calmodulin-dependent kinase II [CaMKII]) or on other subunits (e.g., in phosphorylase kinase [PhK]). A key aspect of regulation recognized in recent years is that many protein kinases are phosphorylated on a residue(s) located in a particular segment in the center of the kinase domain, which is termed the “activation segment.”
This review will focus on the role of the activation segment in the mediation of these different aspects of control. The activation segment is defined as the region spanning conserved sequences DFG and APE and corresponds to residues 184–208 in cAPK. The activation segment includes Thr-197, one of two autophosphorylation sites in cAPK. The conversion of an inactive kinase to an active kinase involves conformational changes in the protein that lead to the correct disposition of substrate binding and catalytic groups and relief of steric blocking to allow access of substrates to the catalytic site. The activation segment and the control of its conformation via phosphorylation plays a key role in these transformations. It can be involved in recognition of regulatory subunits, in autoinhibition of substrate binding, and in promotion of the correct orientation of domains and catalytic site residues. This review summarizes our current understanding of control by the activation segment based on recent structure determinations of active and inactive kinases.
The first observation of Thr-197 phosphorylation in the activation segment of cAPK was reported in 1979. Although it was speculated that phosphorylation at discrete sites might be of physiological importance in the regulation of enzyme activity, it was not until 1990 that mutagenesis studies indicated the significance of this site for the recognition of the regulatory subunit, and not until 1993 that it was definitively shown that phosphorylation is promoted by an autocatalytic event that is crucial for activation. The crystal structure determination of cAPK in 1991 showed the structural importance of Thr-197 phosphorylation and demonstrated possible roles of phosphorylation in promotion of activation. The structure provided a definitive model to which other kinases could be related. Also in 1991, both the fission yeast cell division control kinase (cdc2) and the microtubule-associated protein kinase (MAPK) were found to be activated by phosphorylation on residues that mapped to a position similar to Thr-197 in cAPK. These results showed the importance of this site not only as an autophosphorylation site, as in cAPK, but also as a site involved in kinase cascade activation mechanisms. For the tyrosine kinases, autophosphorylation of pp60v-src at position Tyr-416 (now known to be in the activation segment) had been shown in the early 1980s, and its significance for control in the cellular counterpart of Src kinase was established by 1987. The following year, trans-autophosphorylation of the insulin receptor tyrosine kinase (IRK) was elaborated, and the similarity in sequence location of some of these sites to that in Src kinase and its relatives and in cAPK was noted. As more and more kinases have been discovered and sequenced and further kinase cascades established, it is recognized that control by phosphorylation in the activation segment is a property of most, but not all, protein kinases.
Kinase Structures
The crystal structures of eight unique protein kinases have been reported. Three of the protein kinases have been crystallized in active conformations (cAPK, PhK, and CK1), one in a partially active conformation (CDK2 in complex with cyclin A), and five in inactive conformations (CDK2, MAPK, IRK, twitchin kinase, and CaMKI). Five of these kinases are regulated by phosphorylation on residues in the activation segment (cAPK, CDK2, MAPK, IRK, and CaMKI) and three are not (PhK, CK1, and twitchin kinase).
Protein Kinases Crystallized in Active Conformations: cAPK, PhK, and CK1
The first X-ray study of a protein kinase, cAPK, revealed the basic architecture that has been observed in all subsequent kinase domain structures. cAPK is a key player in cellular responses to the second messenger cyclic AMP. The inactive form is a heterotetramer of two regulatory and two catalytic subunits. Activation is mediated by binding of cyclic AMP to the regulatory subunits, which causes the release of the catalytic subunits. cAPK is primarily a cytoplasmic protein, but upon activation it can migrate to the nucleus, where it phosphorylates proteins important for gene regulation.
The first cAPK structure was a binary complex with a 20 amino acid pseudosubstrate inhibitor peptide. Later, structures of the ternary complex of cAPK with inhibitor and either Mg2+/ATP or Mn2+/AMP-PNP were also solved. The kinase core comprises a bilobal scaffold that has an N-terminal lobe composed almost entirely of β sheet (colored white in Figure 1) and a C-terminal lobe in which α helices dominate (colored pink in Figure 1). The two lobes in the core are joined by a polypeptide chain, which functions as a hinge (magenta in Figure 1), allowing the two lobes to articulate. In the catalytic subunit of cAPK, there are additional residues at the N-terminus and C-terminus that are important for stability, but which show variation in other protein kinases (not shown in Figure 1). The catalytic site is at the interface of the two lobes. The ATP-binding site spans both lobes. The inhibitor peptide (colored green in Figure 1) is associated mostly with the C-terminal lobe. In the binary complex with inhibitor peptide or in the ternary complexes, the lobes adopt a closed conformation. However, in the structure of apo cAPK or cAPK in a binary complex with an iodinated inhibitor, a more open lobe orientation was observed, supporting results from solution studies that had also identified open and closed conformation. Such transitions seem likely to be important: the open form is necessary to allow access of ATP to the catalytic site and release of product ADP; the closed form is necessary to bring residues into the correct conformation to promote catalysis.
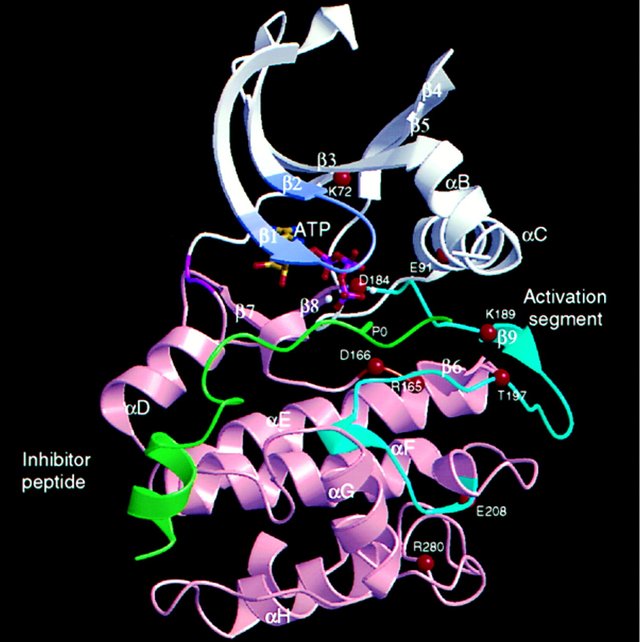
Figure 1
The N-terminal lobe is colored white, and the C-terminal lobe is pink. The hinge region between the lobes is magenta. The inhibitor peptide is shown in green with the position of the serine that is the target for phosphorylation shown as P0. ATP is in ball and stick representation. The glycine-rich region that is important for localization of the phosphates of ATP is shown colored light purple. α Helices and β strands are labeled. The diagram is rotated about 45° to the conventional view to display the activation segment (residues 184–208), which is colored cyan and includes strand β9. The Cα positions of other important residues are labeled and are referred to in the text.
The positions of some of the residues important for binding ATP in cAPK are shown as red circles in Figure 1, and their roles are illustrated in Figure 2. These are conserved in other kinases. The correct alignment of the ATP with respect to the catalytic residues appears crucial for catalysis. The adenine is located in a hydrophobic pocket, and the ribose is stabilized by hydrogen bonds. The phosphates of ATP are aligned for catalysis by interactions with the main chain nitrogens of a glycine-rich loop (light purple in Figure 1), by interactions with Lys-72 (which is localized by a salt bridge to Glu-91), and by interactions with Mg2+ ions (white spheres). One Mg2+, identified as the activatory Mg+, binds to the β and γ phosphates of ATP and to Asp-184 (Figure 2). The catalytic aspartate, Asp-166, which is invariant in all kinases, is located in the C-terminal lobe in a loop, termed the catalytic loop (orange in Figure 1). The aspartate is presumed to act as a base to remove a proton from the protein substrate hydroxyl group (Figure 2), although the catalytic mechanism is not definitively established. The resulting alcoholate or phenolate ion is positioned to attack the γ phosphate of ATP.
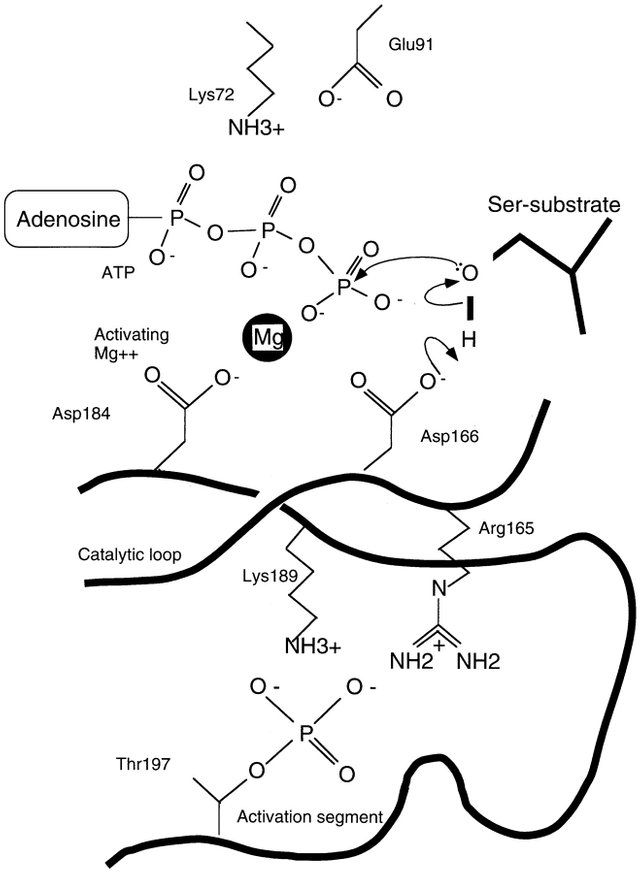
Figure 2
A possible mechanism is shown in which base-catalyzed attack by Asp-166 on the substrate serine promotes the formation of an alcoholate ion, which can then attack the γ phosphate of ATP. The catalytic mechanism is not definitively established.
The activation segment (cyan in Figure 1) begins at the highly conserved DFG motif with Asp-184. The segment continues with a region that is remarkably different in the active and inactive kinase structures. In cAPK there is a short β strand, β9, which is followed by a loop containing the autophosphorylation site Thr-197. The segment ends in the region that has a 310 helix structure and includes residues that are important for the peptide substrate hydrophobic recognition site C-terminal to the phosphorylated serine (labeled P0 in Figure 1). The final residue, Glu-208, is part of the APE motif, which is conserved in most kinases, and is hydrogen bonded to the conserved arginine Arg-280.
As shown in Figure 3a the phosphothreonine Thr-197-P contacts His-87 from the N-terminal lobe, Arg-165, which precedes the catalytic base (Asp-166), Lys-189, and Thr-195. The phosphate is placed to compensate the cluster of positively charged residues. Steady-state kinetic analysis combined with viscosometric measurements of cAPK mutants, in which the threonine was changed to an aspartate or an alanine, showed reductions in catalytic efficiency that were due to the specific effects of weakened ATP affinity and reduced rates of phosphoryl transfer. There was little effect on peptide substrate binding affinities. These results imply that the Thr-197-P group is important for modulating the catalytic flux of cAPK.

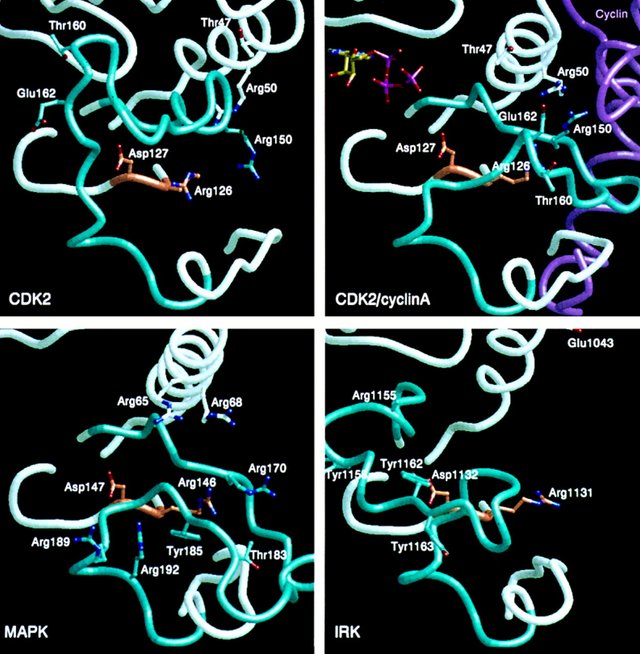
Figure 3
The view is similar to that in Figure 1. The activation segment is colored cyan, the catalytic loop orange, and the remainder of the polypeptide chain white. The inhibitor peptide in (a) for cAPK is shown in green. The kinases were superimposed using coordinates for the C-terminal lobe corresponding to residues 128–253 in cAPK. The fragments displayed correspond to cAPK residues 43–100 (including the glycine loop that interacts with ATP and the C helix), residues 164–171 (the catalytic loop), and residues 183–218 (including 184–208, the activation segment). (a)–(d) represent four active kinase structures; (e), (g), and (h) represent inactive kinase structures, and (f) is a partially active kinase.
(a) cAPK in the active, closed, and phosphorylated state. Thr-197-P interacts with His-87, Lys-189, and Arg-165. Arg-165 precedes Asp-166, the presumed catalytic base.
(b) PhK in the constitutively active state. Glu-182 occupies a similar position to Thr-197-P in cAPK and interacts with the arginine, Arg-148, adjacent to the catalytic base. Lys-72 is over 5 Å from the glutamate.
(c) CK1 in the active state. A sulfate ion is bound in the crystalline protein and contacts Lys-159, Lys-176, and Arg-130.
(d) CK1 (mammalian) in the active state. Asn-177 occupies a similar position to Thr-197-P in cAPK and interacts with Arg-130. Arg-130 also interacts with main chain carbonyl oxygens from the activation segment.
(e) CDK2 in the inactive and closed state. Thr-160 is close to the glycine loop at the ATP-binding site.
(f) CDK2–cyclin A in a partially active conformation. Cyclin A is shown in magenta. Both the C helix (containing the PSTAIRE motif, residues 45–51) and the activation loop shift significantly from their positions in inactive CDK2. Glu-162 occupies a position approximately equivalent to His-87 in cAPK and hydrogen bonds to Arg-126, the arginine adjacent to the catalytic base, and Arg-150. Arg-50 and Arg-150 also interact with cyclin A.
(g) MAPK in the inactive and open conformation. Thr-183 is exposed, and Tyr-185 is buried. Arg-65, Arg-68, Arg-170, and Arg-146 could form a recognition site for a phosphorylated residue in the active state. Arg-189 and Arg-192 form a second dianion recognition site.
(h) IRK in inactive and open conformation. The activation segment blocks the catalytic site, and Tyr-1162 is hydrogen bonded to Asp-1132, the catalytic aspartate. Arg-1131 and Arg-1155 could form a potential phosphate recognition site in the active state after substantial conformational change.
Comparison of Inactive and Partially Active Kinase: CDK2 and CDK2–Cyclin A Complex
The determination of the crystal structures of the inactive human CDK2 and the partially activated human CDK2–cyclin A complex has provided remarkable insights into the activation mechanism of the CDKs. The CDKs are regulatory enzymes that initiate and coordinate events of the eukaryotic cell cycle. Their role in cell proliferation is determined by elaborate control mechanisms. The activity of CDKs is dependent upon their association with a cognate cyclin, whose concentration varies during the cell cycle. The CDK–cyclin complex may exhibit some kinase activity, but requires phosphorylation of a threonine residue (Thr-160 in human CDK2) for full activity. The crystal structure of CDK2 in its inactive state provided an image of an inactive kinase. The basic closed bilobal architecture is similar to cAPK, but there are several major differences in structure between inactive CDK2 and active cAPK. The activation segment, containing the phosphorylatable threonine Thr-160, is folded so that it blocks the substrate recognition site (Figure 3e). The helix corresponding to helix C in cAPK (see Figure 1) contains the PSTAIRE sequence (residues 45–51) of CDK2 and is known to be important for recognition of cyclin A. This helix is displaced in CDK2, and residues important for binding ATP are wrongly disposed.
Inactive Kinase Structures: MAPK, IRK, Twitchin Kinase, and CaMKI
The inactive kinase structures show four very different conformations of the activation segment, indicating that there are a variety of conformations accessible to different kinases in the inactive state.
MAPK is phosphorylated on two residues in the activation segment, Thr-183 and Tyr-185. Following the discovery of MAPK as a crucial component in the response to insulin, subsequent work showed that several protein kinases phosphorylated on tyrosine residues in response to mitogens and certain transforming agents were identical or closely related to MAPK and established a central role for this kinase in signal transduction pathways.
IRK exhibits a dramatic example of autoinhibition by the activation segment. The insulin receptor is an α2β2 heterodimer. The extracellular α chain is responsible for binding insulin, and the intracellular part of the β chain contains the tyrosine kinase domain, which is flanked by a juxtamembrane region and a C-terminal tail, both of which are targets for phosphorylation. The primary substrate of the insulin receptor is the receptor itself. The autophosphorylation of three tyrosines (Tyr-1158, Tyr-1162, and Tyr-1163) in the activation segment of the kinase domain results in activation.
Twitchin kinase reveals a further twist in interactions that stabilize the activation segment. Twitchin, the unc-22 product that is involved in muscle function, is a 753 kDa protein composed of multiple copies of both fibronectin type III–like domains and immunoglobulin-like domains and a Ser/Thr protein kinase, which is located near the C-terminus. The kinase portion exhibits 52% identity to smooth muscle MLCK, and, like MLCK, twitchin kinase is regulated by autoinhibition by 60 residues C-terminal to the kinase core.
The recently determined structure of CaMKI has provided a further elaboration of the autoinhibitory mechanism. CaMKI phosphorylates the synaptic vesicle–associated proteins synapsin 1 and 2, thereby modulating interactions of synapsin with actin filaments and neurofilaments. The structure of CaMKI in the absence of Ca2+/calmodulin reveals an open conformation in which there are extensive interactions between the C-terminal autoinhibitory sequence and the kinase core. However, the autoinhibitory region does not enter the ATP-binding site (as it does in twitchin kinase), but interacts on the outside of the ATP-binding domain, leading to conformational changes at the ATP-binding site. The activation segment is disordered. Conversion to the fully active CaMKI requires both binding of Ca2+/calmodulin and phosphorylation on a threonine in the activation segment by a specific kinase.
Discussion
The structures of the kinases in the active conformation all show equivalent positions for essential catalytic site residues Lys-72, Asp-166, and Asp-184 in cAPK. Their positions and the correct orientation of the Mg2+/ATP and the protein substrate appear crucial for catalysis and are dependent upon the tertiary structure of both lobes and the correct relative orientation of the lobes. Both CDK2 and MAPK bind ATP in their inactive conformations in the crystal, but the γ phosphate is either in the wrong position to promote in-line transfer to the substrate or not located, thus demonstrating the subtle dependence of activity on structure. In the inactive IRK and CaMKI structures, the ATP site is blocked or destroyed.
The activation segments of protein kinases vary in length (up to 10 amino acids). The variability in sequence may allow the kinase to be constitutively active, or it may allow control by autophosphorylation, if the segment has a sequence corresponding to the substrate specificity of the kinase itself, or control by phosphorylation directed by other kinases that function as part of a cascade. The structural studies provide examples of each of these mechanisms, but there are other possibilities. For example, it is possible that a phosphate recognition site generated and normally recognized by a phosphorylated residue from the activation segment could also bind a phosphoresidue from elsewhere in the molecule, and this could provide a further inhibitory mechanism. More definite information is required to understand the exquisite regulatory properties of this enormous family of enzymes, for which phosphorylation on the activation segment is only one aspect of a number of diverse schemes for regulation.
Go here https://steemit.com/@a-a-a to get your post resteemed to over 72,000 followers.